4. KAT Walkthrough¶
KAT is a multi-purpose toolkit and even we, the authors, have probably not fully mined all the possible scenarios in which the software can be applied. This section however lists some typical applications for which it has been used.
4.1. Comparing R1 v R2 in an Illumina PE dataset¶
Comparing the k-mer spectra of read 1 to read 2 files of a paired-end (PE) sequencing run gives you several insights into properties such as shared errors, difference in quality, and provides a decent benchmark to compare two similar runs. Here’s how to do it:
kat comp -t 16 -n -o <output_prefix> <R1.fastq> <R2.fastq>
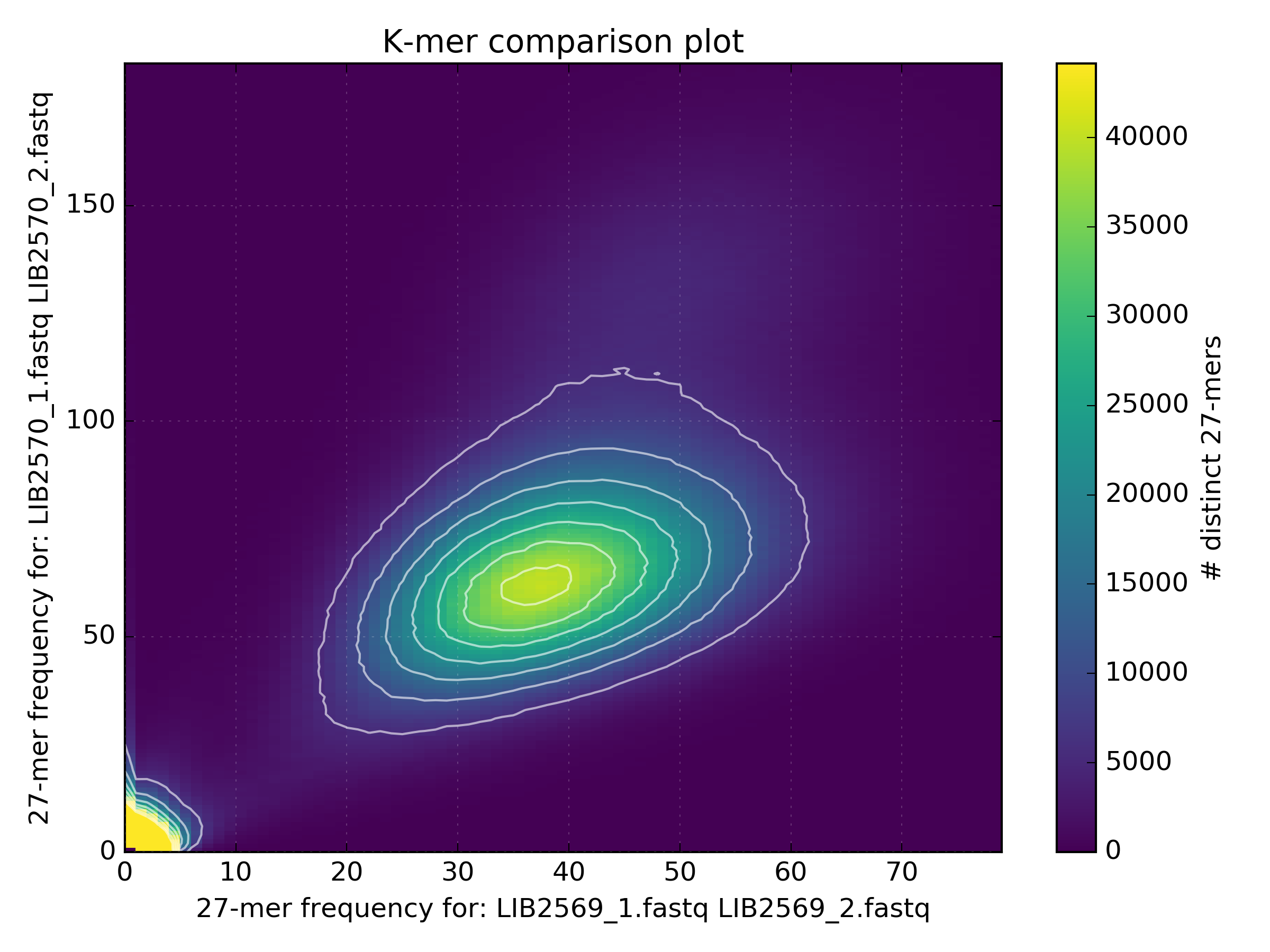
Executing this command counts each fastq file into separate k-mer hashes, then creates a matrix of k-mer spectra frequencies in each dataset for each distinct k-mer. Finally, a density plot is made of the matrix. There are a number of interesting points in the output of the run. First, the some basic stats are produced on the standard output. Check properties like expected unique size, mean coverages for shared/unshared, or the distance measures between the spectra. These values are just indicative, but might point to disaster ahead. The density plot also provides a visual representation of how much shared errors are affecting your data. The following plot shows what you would expect to see for a completely unbiased homozygous PE library of S.coelicolor.
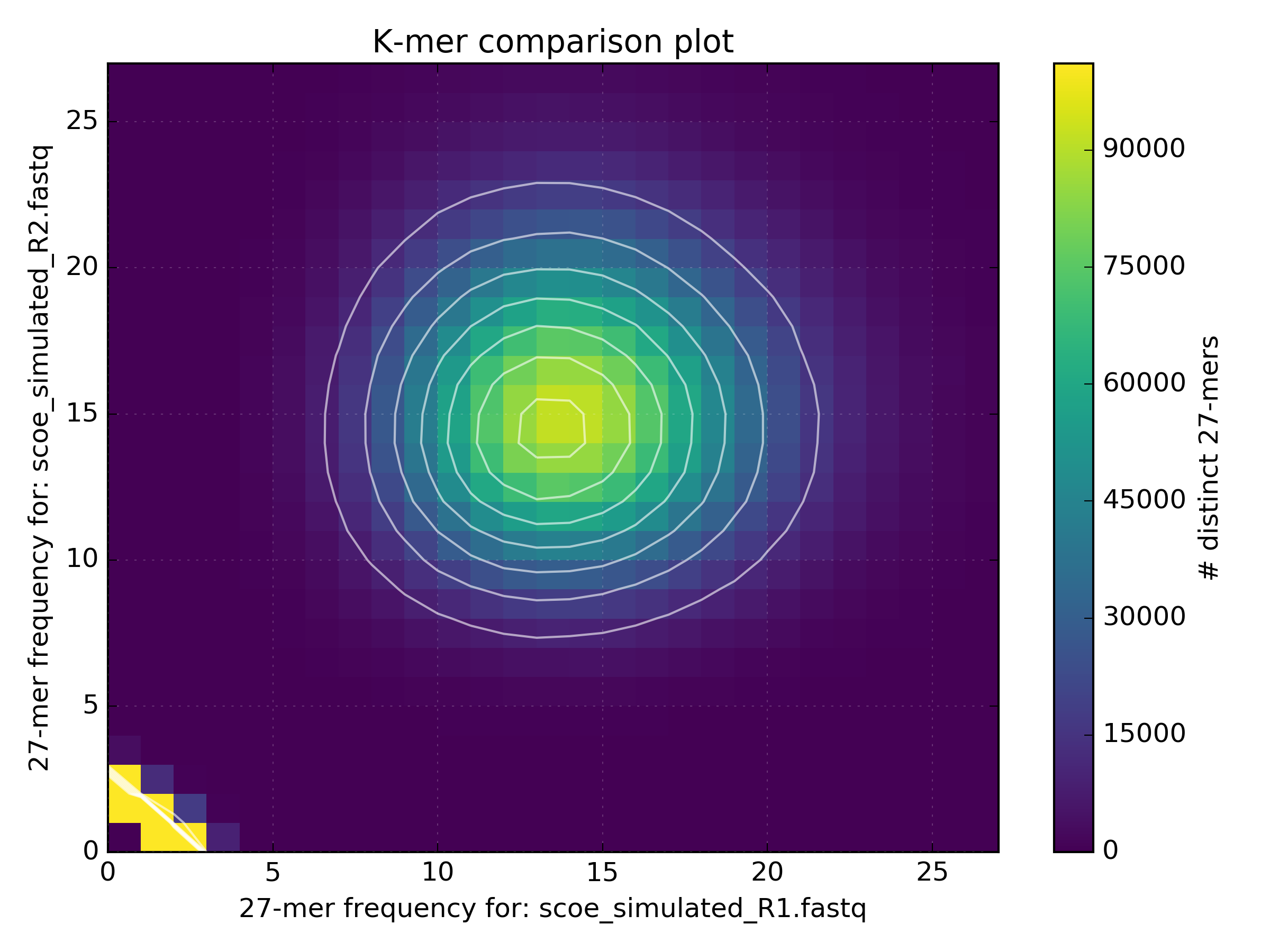
However in reality, various biases can interfere with sequencing experiments and we will probably end up with data which isn’t quite as clean, such as that shown in the following plot from an Illumina run of S.cerevisiae S288C.
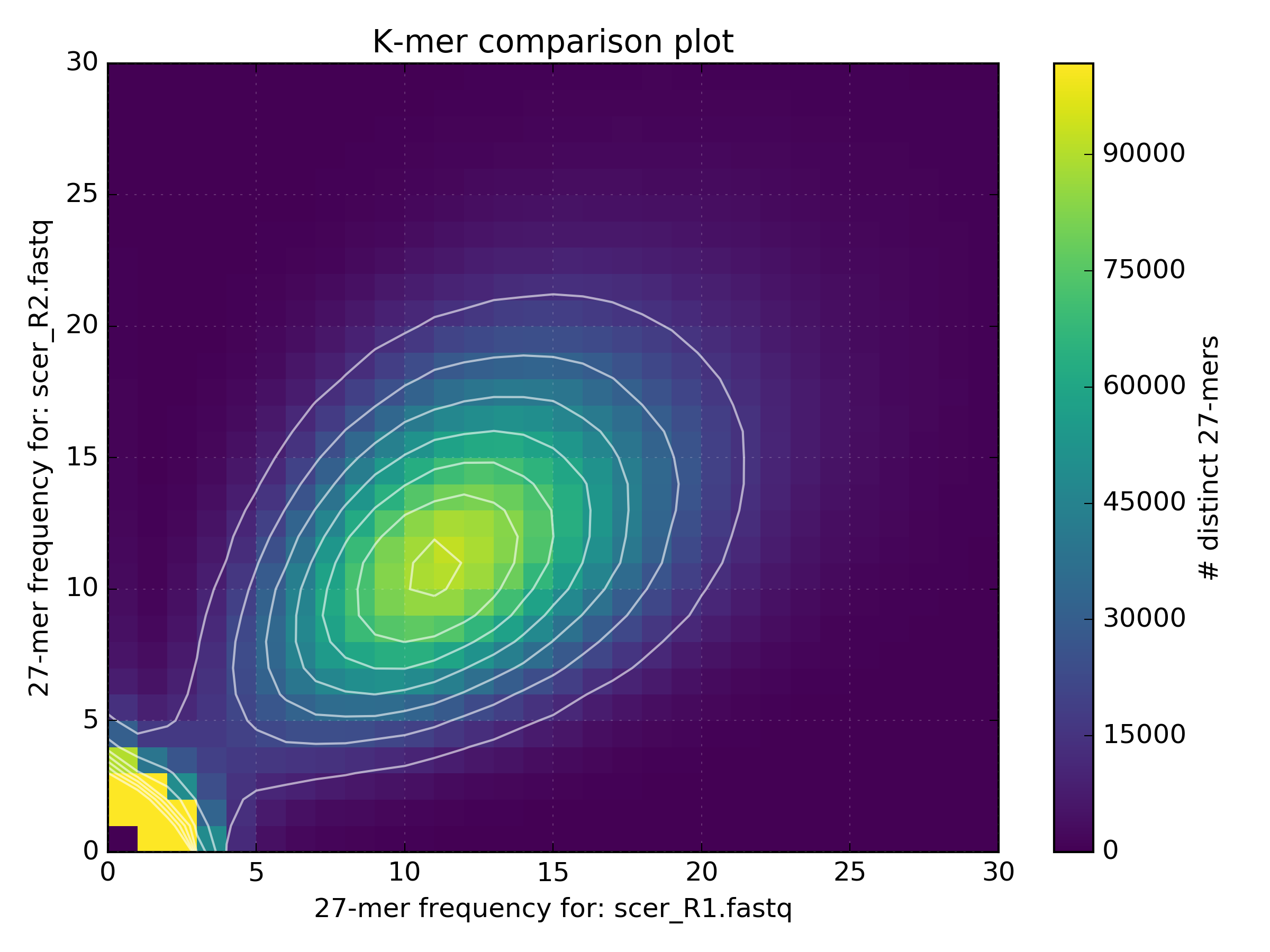
It’s also useful to examine the shared and unique partitioned spectra between R1 and R2. This can be done by using the same matrix file. The plot below is from the same S.cerevisiae S288C dataset, generated using the following command line:
kat plot spectra-mx -i [options] -o <output_file> <matrix>
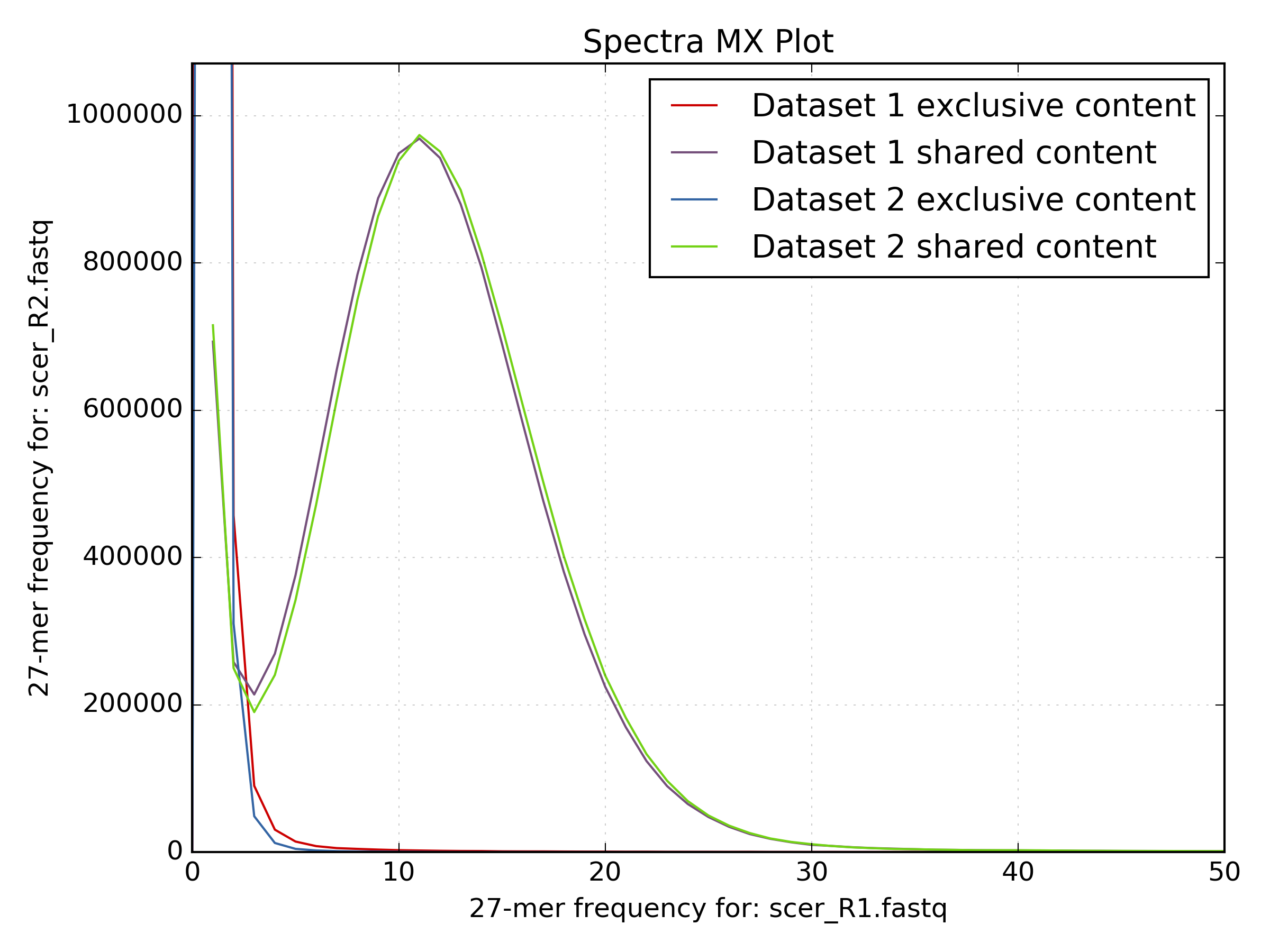
When generating plots, KAT uses input parameters supplied to the tool to generate a plot title and labels for the x- and y-axis. If you want to change these you can regenerate the plot from the matrix using the kat plot tool directly. For example, to regenerate the density plot from the S.coelicolor dataset above you can run the following command after running the kat comp command:
kat plot density -o <output_prefix> -t <title> -a <x-axis label> -b <y-axis label> <matrix>
4.2. Detecting GC bias¶
Using KAT it is possible to correlate biases due to GC content. KAT does this by combining k-mer frequency with the GC count for each distinct k-mer and representing the data in a matrix which can be plotted in a similar way to that discussed in the previous section. The command to produce a matrix of GC counts to k-mer frequency is as follows:
kat gcp -t 16 -o <output_prefix> (<fastq>)+
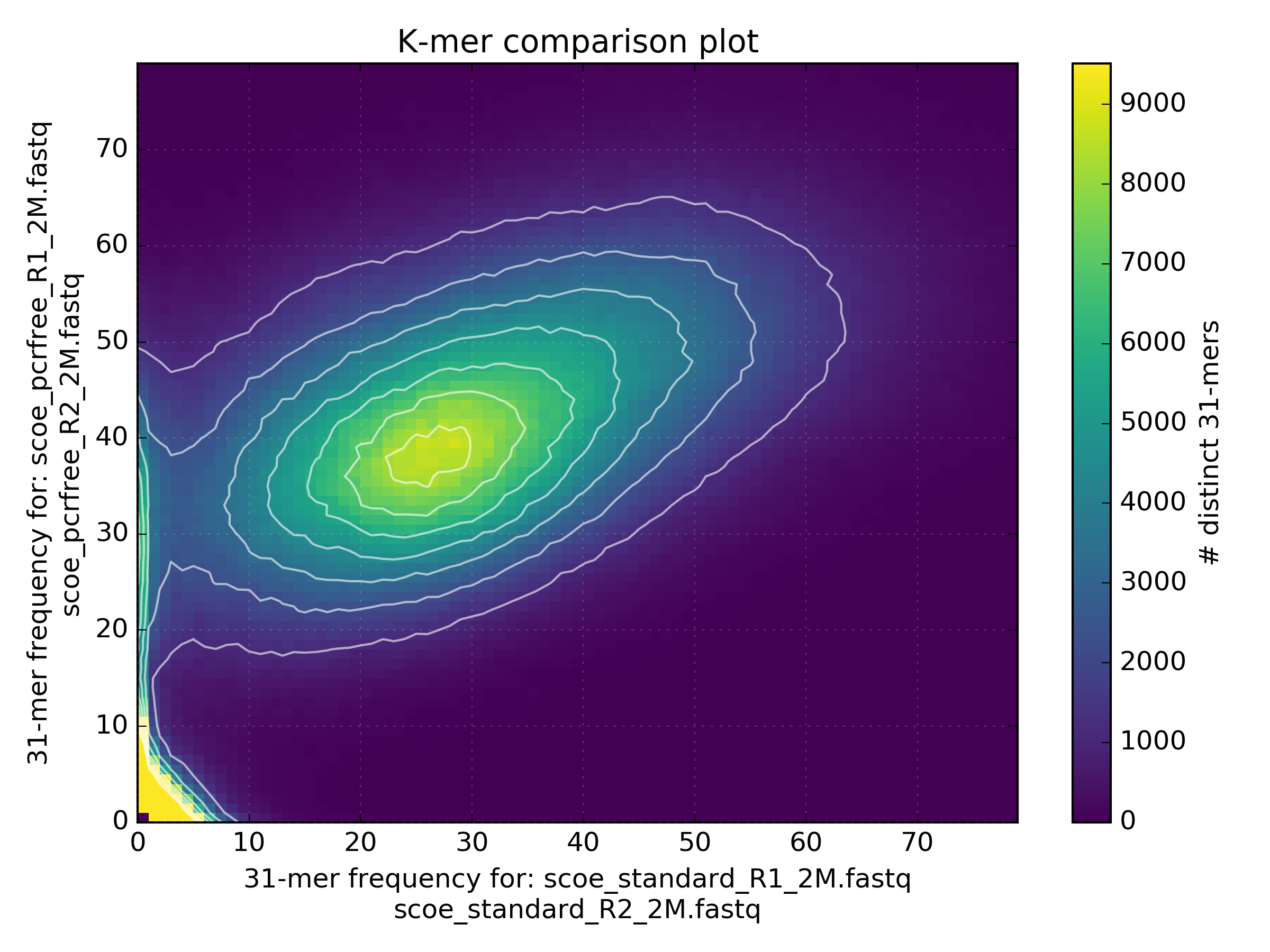
The following figure shows how GC bias varies depending on the protocols used in the sequencing experiments: 1) Simulated, 2) PCR-free, 3) PCR after adaptor ligation, 4) Standard protocol. In the simulated data there is a clean circle positioned at the expected GC levels for S.coelicolor, which has a GC rich genome. GC plots from the other experiments show a less distinct circle with the PCR-free protocol generating the truest reflection of the simulated dataset.
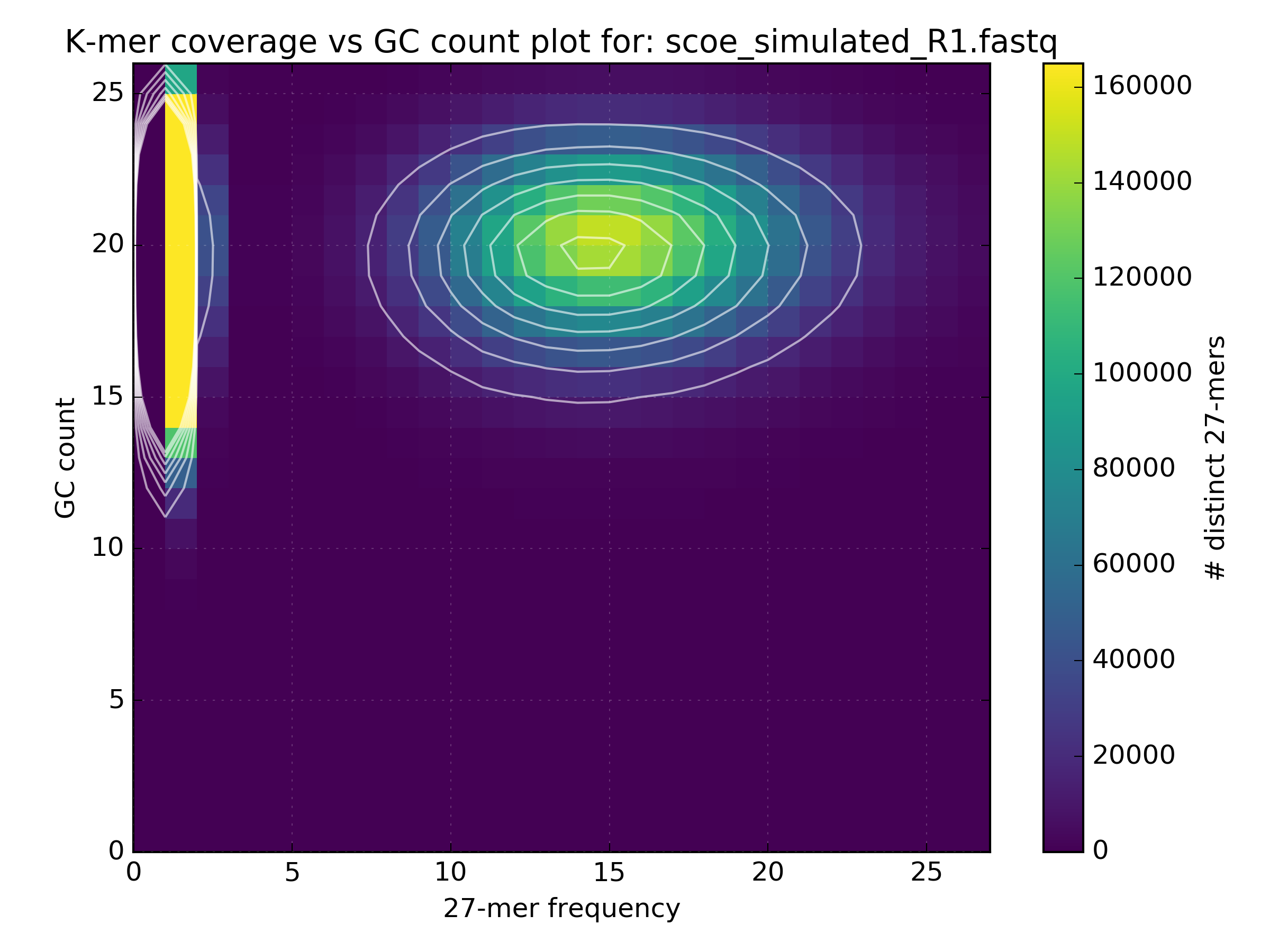
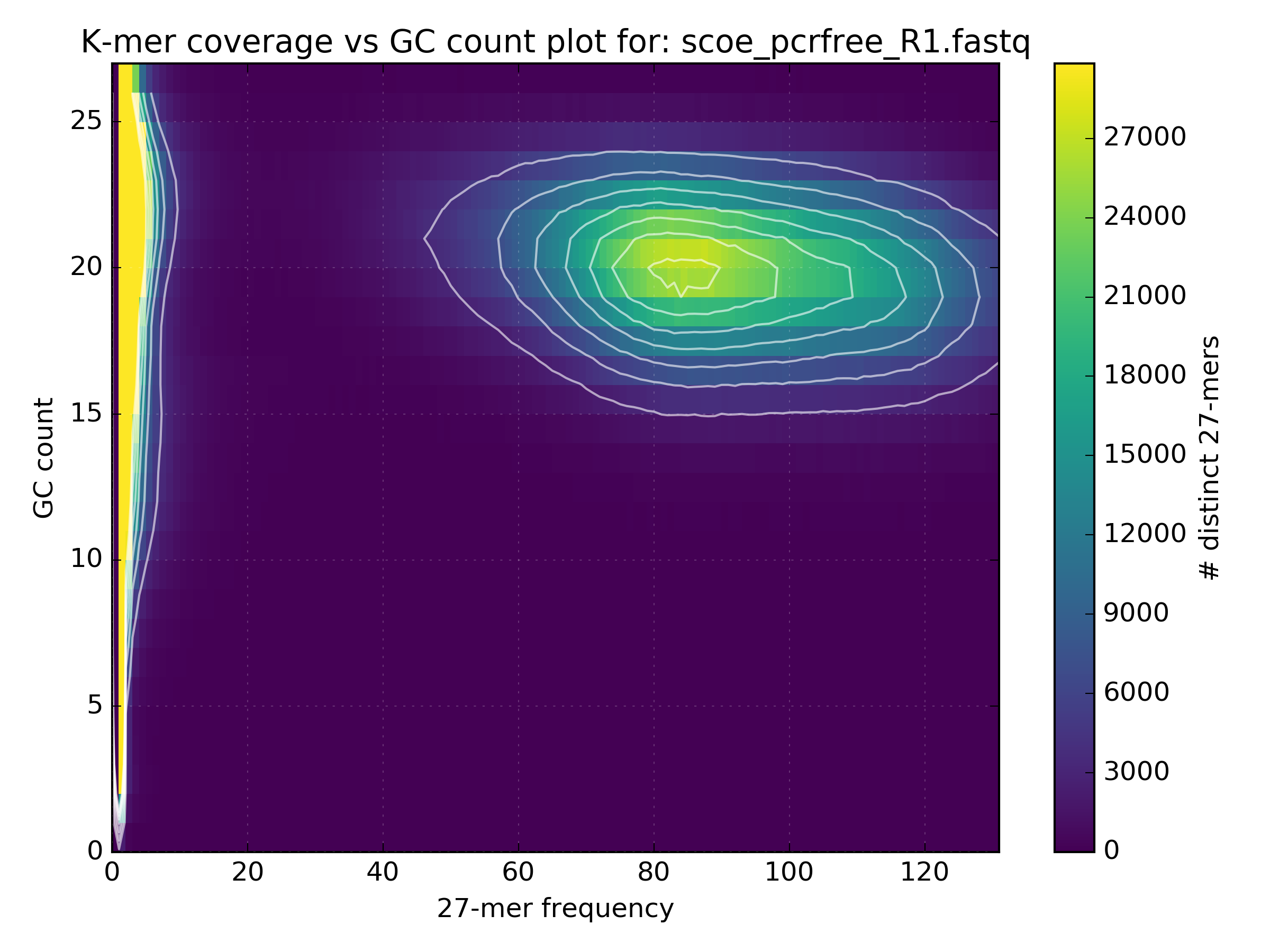
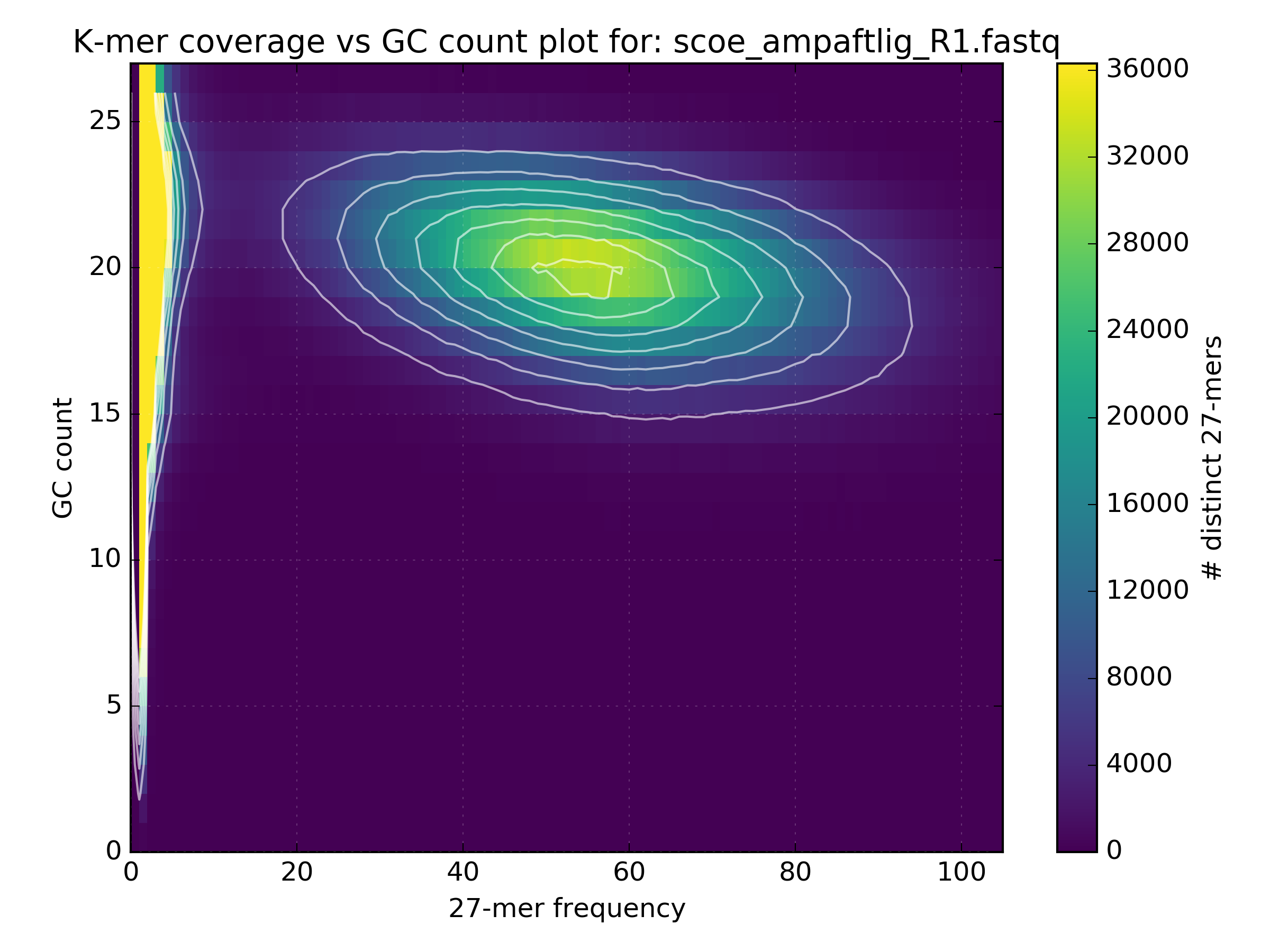
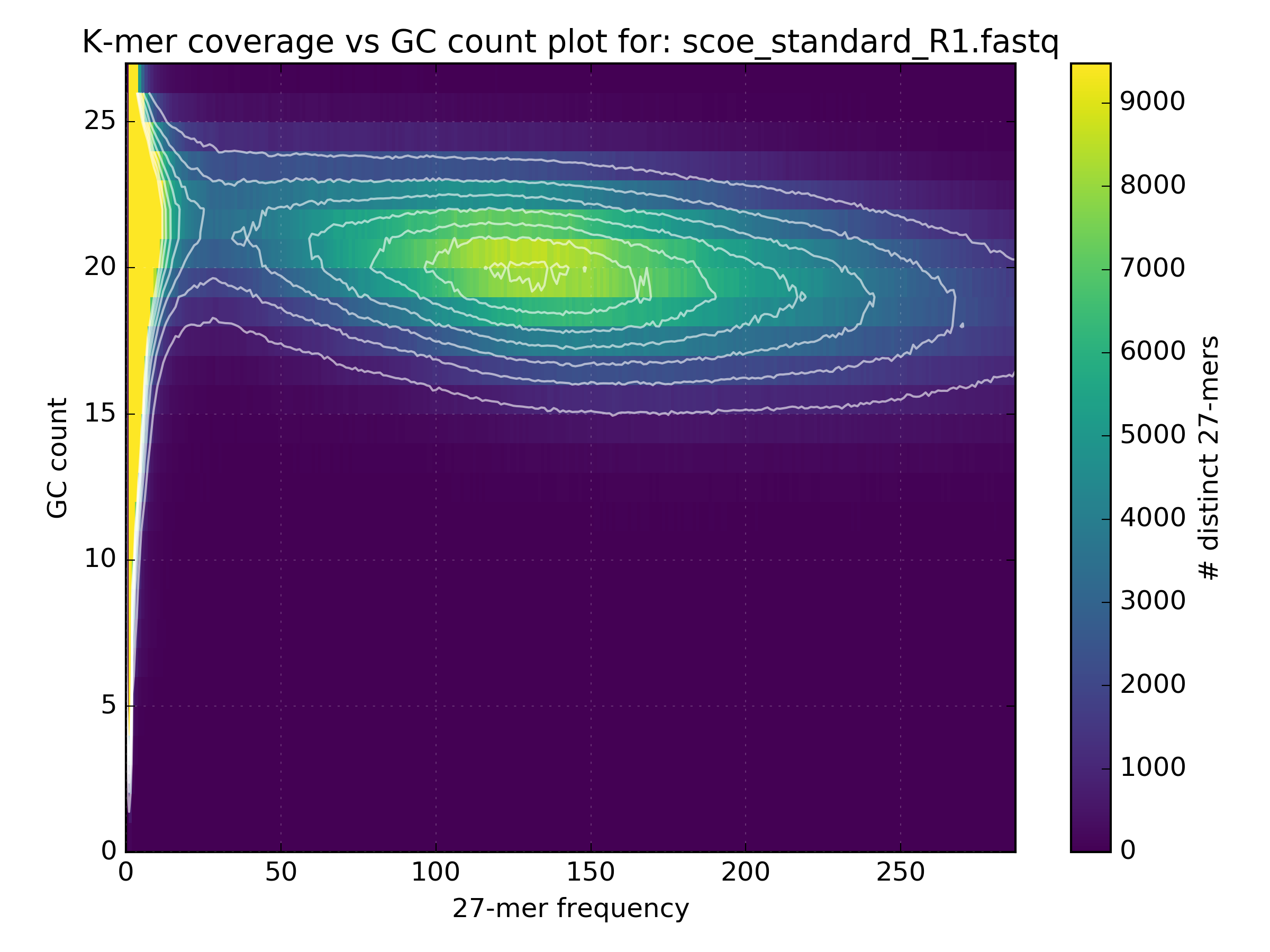
4.3. Checking library consistency¶
In most Whole Genome Shotgun (WGS) projects, you will use more than one library. The most simple case could be one PE library and one Long Mate Pair (LMP) library, but you could also have a dozen libraries of similar or completely different types, for example using different insert sizes or different library preparation protocols. It is good practice to check for obvious incongruence among libraries before trying to assemble them.
When you are sequencing the same genome, you are randomly sampling across it. Therefore you should have different spectrum originating from different experiments, but sampled from the same set. This means that if you decompose your spectra on the components being generated by single-copy elements, duplicated elements, triplicated elements, and so on, every motif belonging to a component distribution should belong to that component distribution across samples.
4.3.1. PE vs PE¶
Paired end sequencing constitutes the bulk of most current efforts on sequencing, and is used as a benchmark to sample the motifs on the genome. Since this data type is expected to have the more random distribution and even coverage, it is a good place to start studying correlation between libraries.
The cleanest examples come from simulated data, where the correlation is virtually perfect as long as belonging to the same distribution, and shows almost no biases. Real data tends to show more correlation within the same distribution, especially in cases where strong biases are in play.
To compare two PE libraries run the following command:
kat comp -n -t 16 -o pcrfree_vs_standard pcr_free 'pcr_free.R?.fastq' 'standard.R?.fastq'
Note that the quotes around the inputs at the end of the command line allow you to group files together into a single input. Therefore all files matching “pcr_free.R?.fastq” are treated as the first input group, and all files matching “standard.R?.fastq” are treated as the second input group. K-mers are counted for each group separately. This saves the user wasting time and disk space concatenating PE files together prior to input into KAT.
The previous command produces only the density plot, so to generate the shared vs unique content plot also run:
kat plot spectra-mx -i [options] -o pcrfree_vs_standard_shared.png <matrix_file>
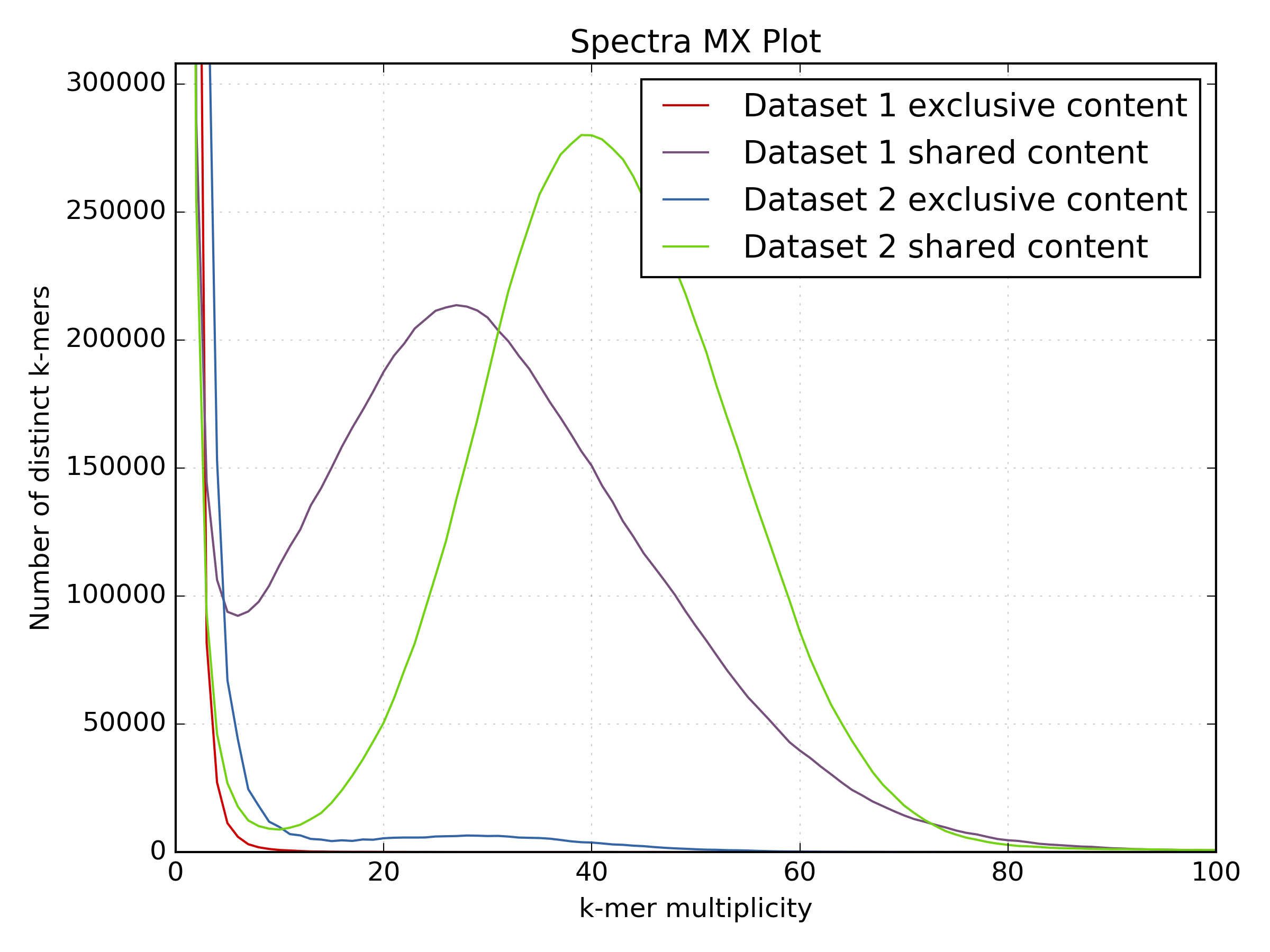
The following plots compare two PE sequencing experiments in C.fraxinea, showing a large motif duplication in one of the experiments. This is obvious from the spectra-mx plot but not so clear in the density plot.
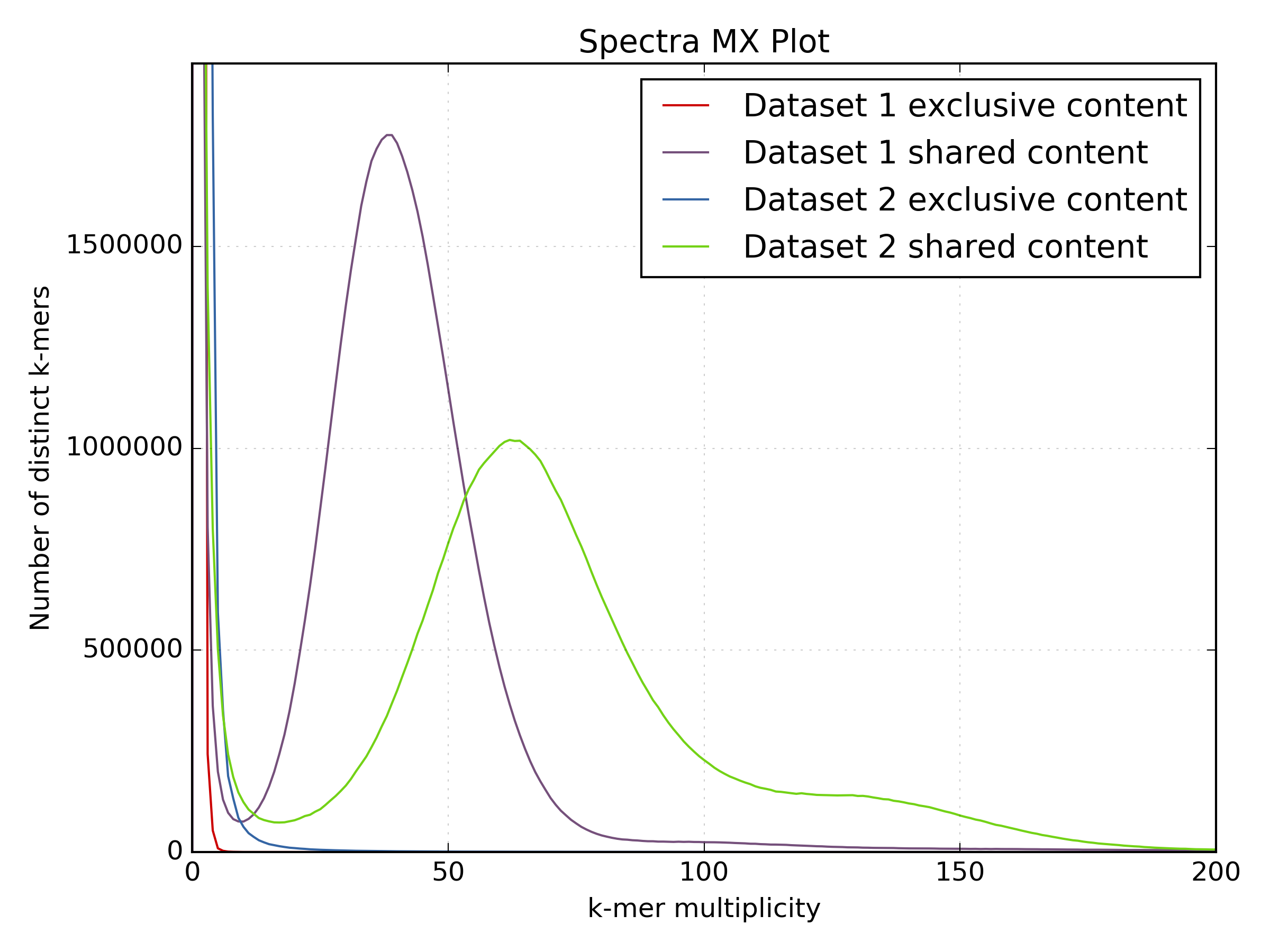
An interesting comparison to perform is between a PCR-free and a Standard protocol using a k-mer spectra density plot. Note that the coverage from the standard protocol is more variable than that generated from the PCR-free protocol. In addition, the blue region at (x=0 y=30) indicates kmers that are sampled by the PCR-free protocol but not the standard protocol. The coverage from the standard protocol is less than from the PCR-free protocol as less sequence was generated from this library.
The following shared content plots generated from the same comparison show the k-mer spectra split on shared and unique content. Note how content is “lost” on the standard protocol as soon as you ask for at least 5x kmer coverage. Although much of this is from the “error distribution” side (where the red and blue lines are truncated at K-mer multiplicity <5), you can also see that real content from the main frequency distribution is being lost by using that cutoff (the increased peak of the blue line around K-mer multiplicity = 30). This should make you think carefully about setting those low-coverage cutoffs again!
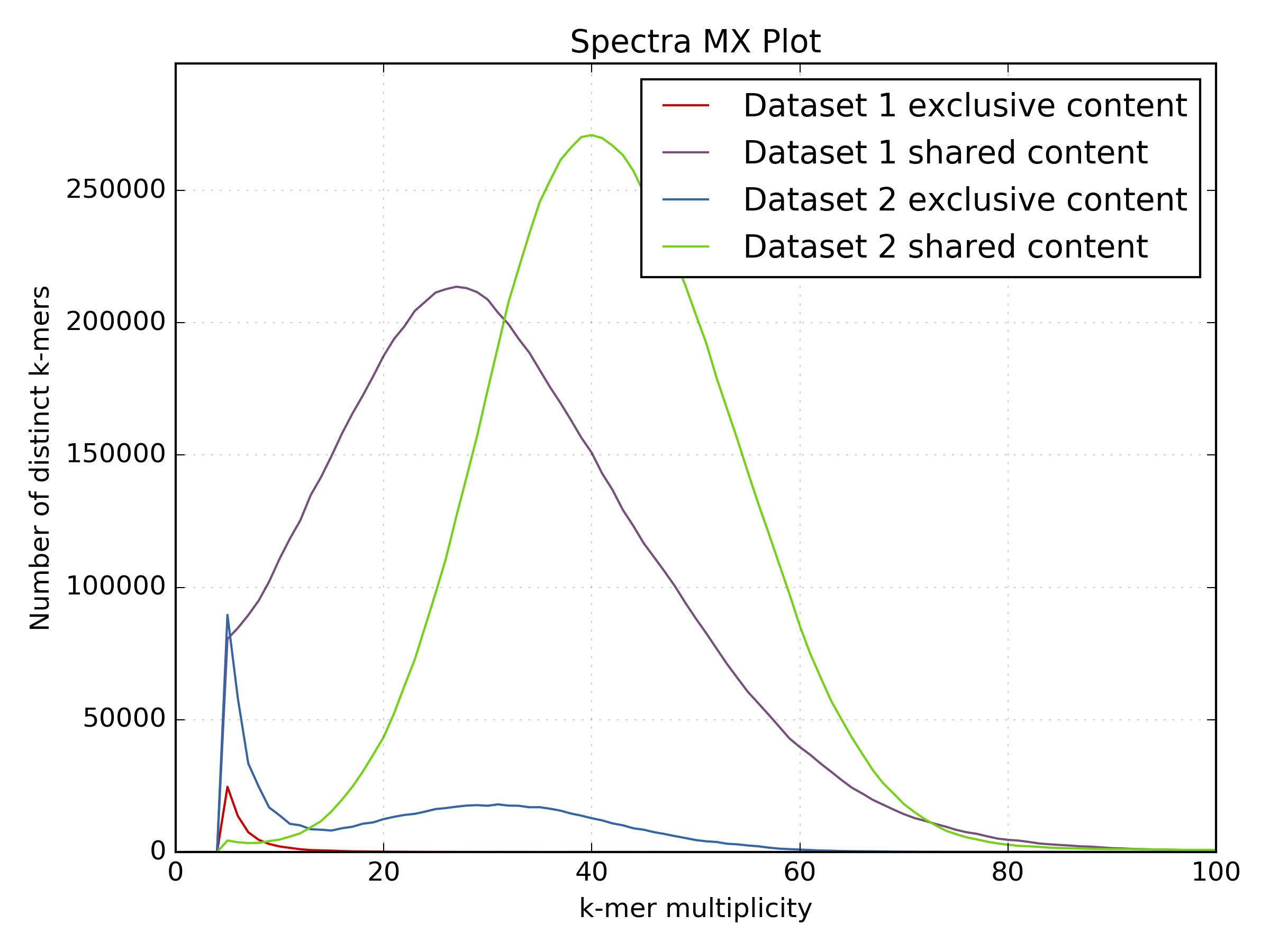
4.3.2. PE vs LMP¶
When using LMP data, in many cases the protocol used to prepare the sequencing library will impose considerable biases. It is good practice to check LMP reads against the PE reads for coherence. They have been prepared from the same genomic DNA so should have similar content. Over-representation and absence of motifs are important factors to check. The presence of motifs originating from adaptors (in fact mostly generated from their junction with genomic DNA) can also be spotted.
In the example shown below, a LMP run is compared to a PE run before processing according to the guidelines for the Nextera LMP protocol:
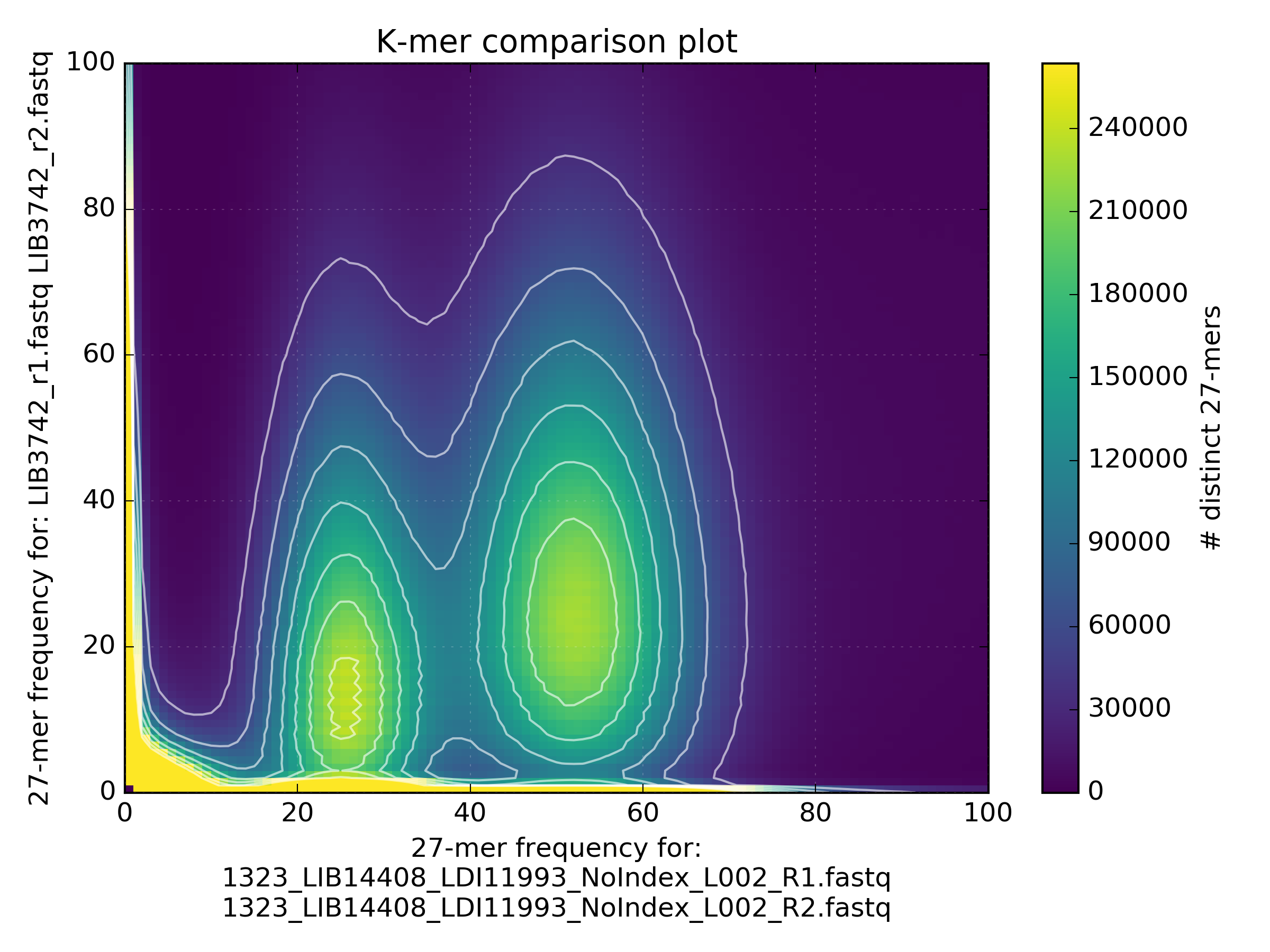
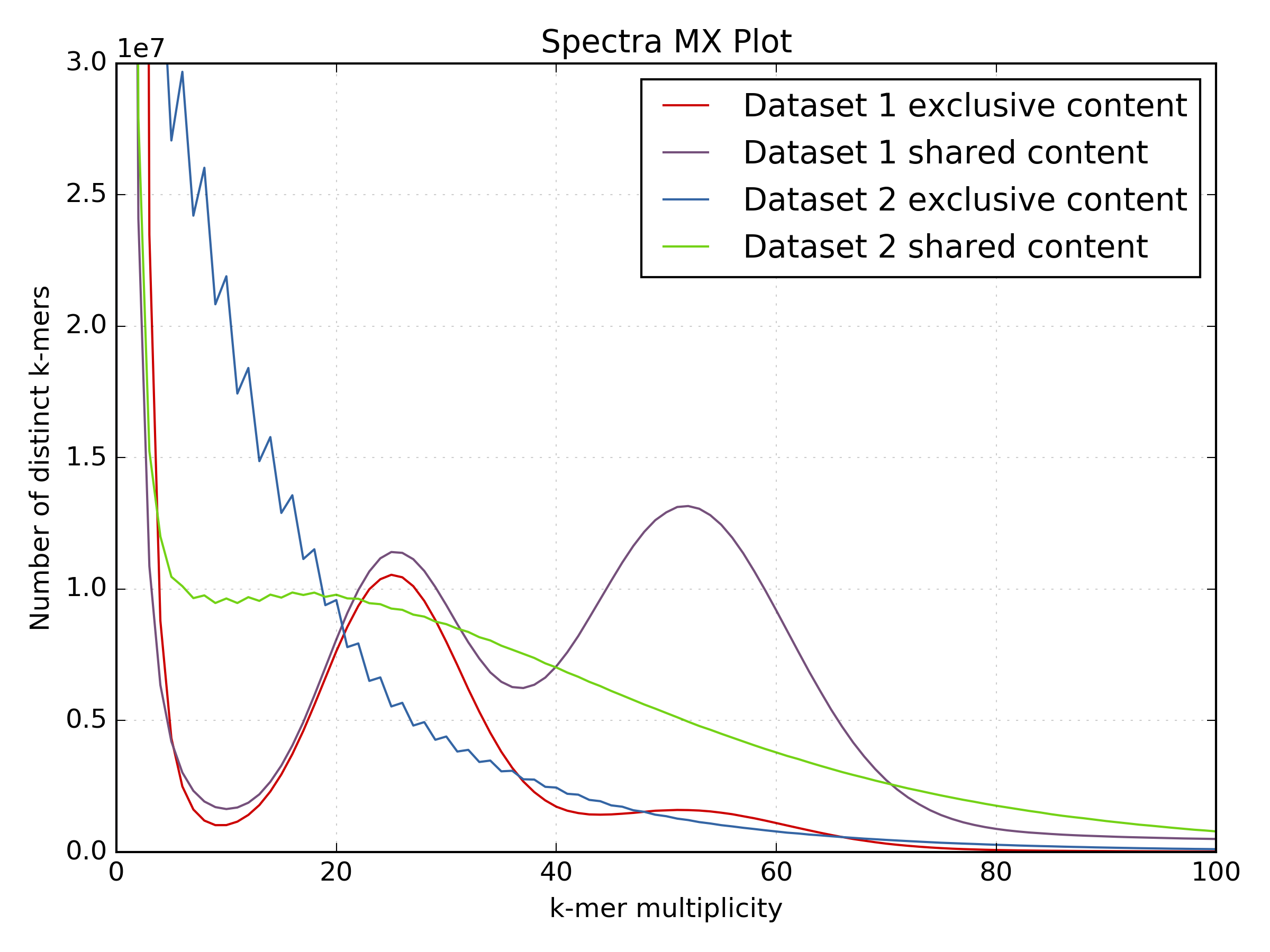
These plots show the same LMP run after processing:
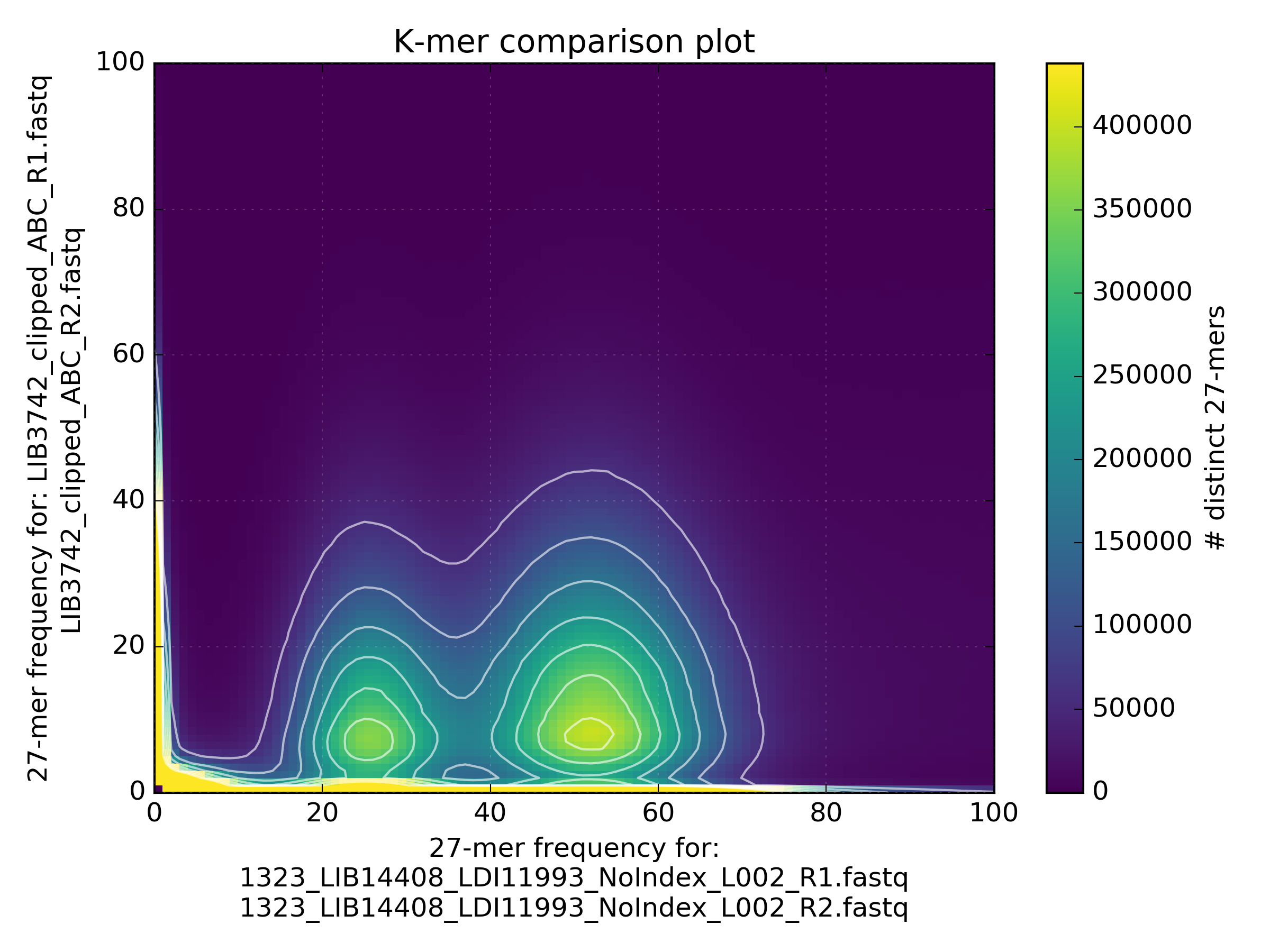
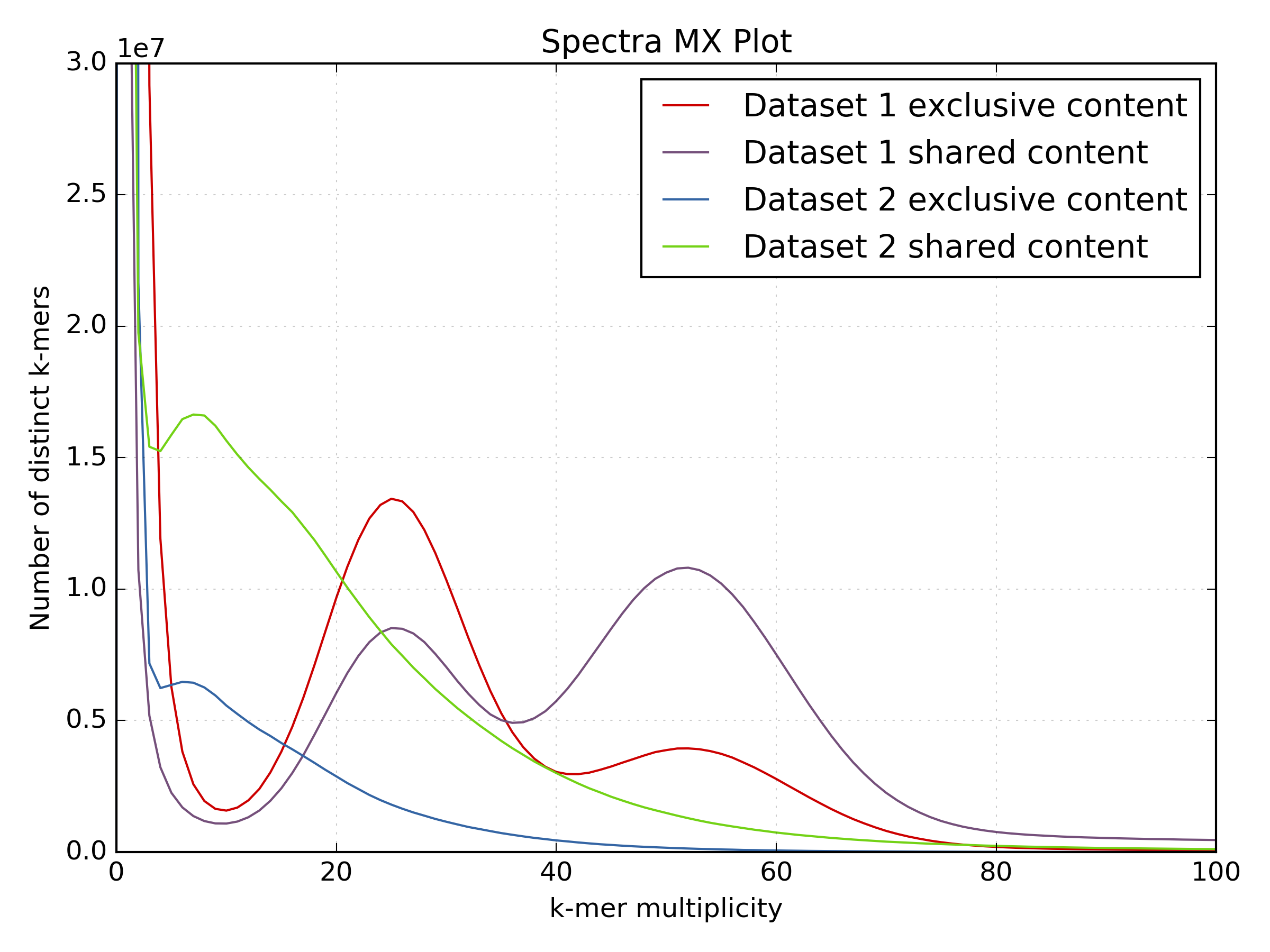
While the motif presence and spectra of the LMP library are certainly better after processing (dataset 2 on the spectra-mx plot), there is content lost and the biases are clearly visible. You can spot representation bias on the density plot for both clusters on teh y-axis. Both clusters are too wide and have large “tails” going up. This is a typical signature for PCR-generated duplications in an early step in the protocol.
Especially interesting is the use of the shared and unique motifs to spot how well the LMP library covers the whole genome. It is usually accepted that for coverages higher than 10 the library should mostly cover the whole genome. If we look at the content “exclusive” to the PE library (the red line) as content not covered by the LMP library, it is obvious that processing the LMP removes a lot of content. While the spectra of the filtered LMP has better distribution, it is clear much content is not there. In this case, the library will not be very useful for scaffolding.
4.4. Contamination detection and extraction¶
Breaking WGS data into k-mers provides a nice way of identifying contamination, organelles or otherwise unexpected content, in your reads or assemblies. This section will walk you through how you might be able to identify and extract contamination in your data.
4.4.1. In reads¶
Detecting contamination in your WGS datasets are reliant on the contamination having differing levels of coverage and/or GC content from your target species. KAT can be used to identify this:
kat gcp [options] (<fastq>)+
Running this tool will produce a matrix containing distinct k-mer counts at varying frequency and GC value. It will also produce a density plot, such as the one below that highlights error k-mers shown at very low coverage with a wide GC spread and genuine content between 10-100X with GC spread from 5-25. In this case we also have some unexpected content shown at approx 200X with GC from 15-25:
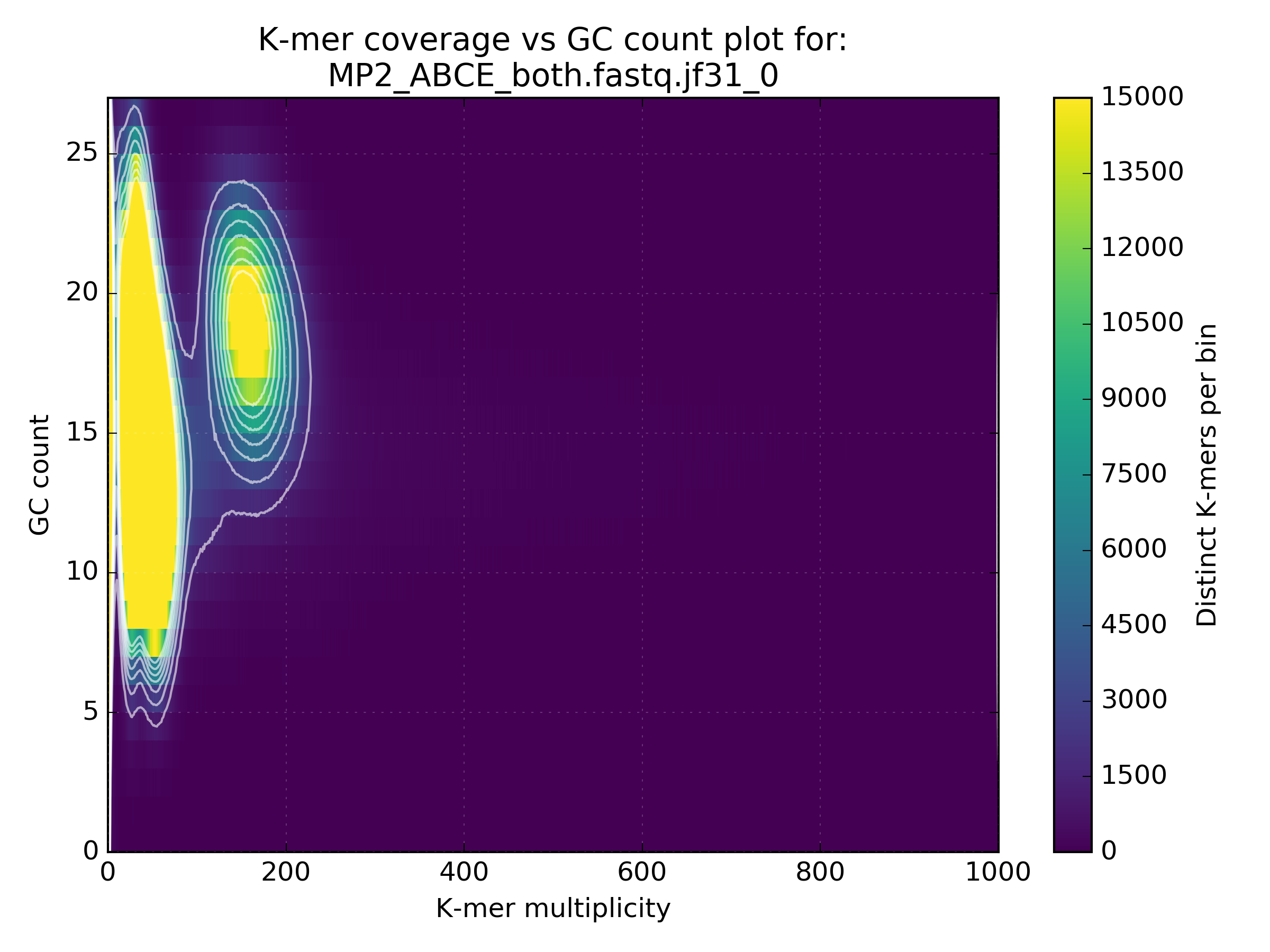
The high coverage hot-spot is already suspicious but it becomes even more so after looking at other WGS libraries of the same sample:
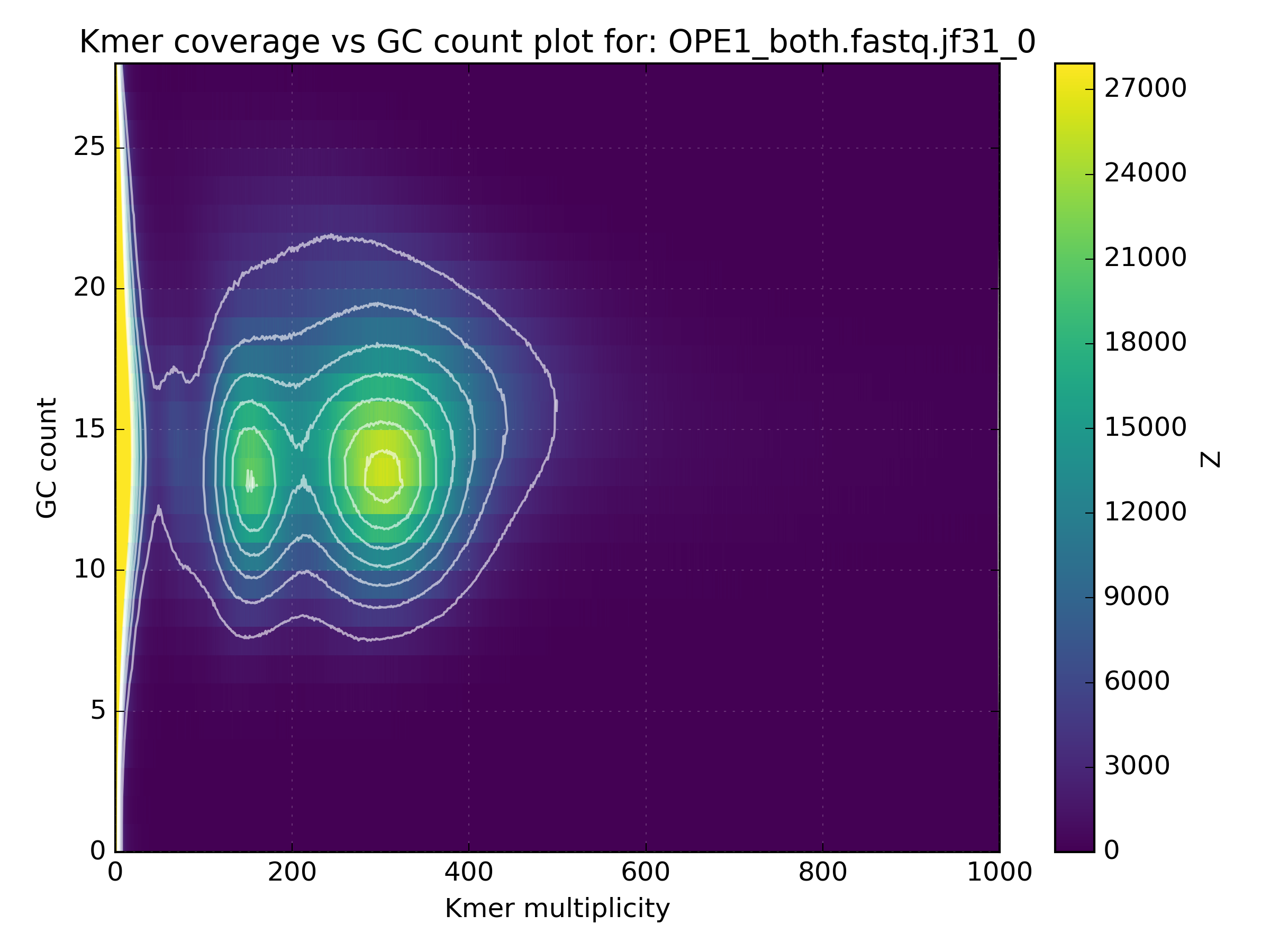
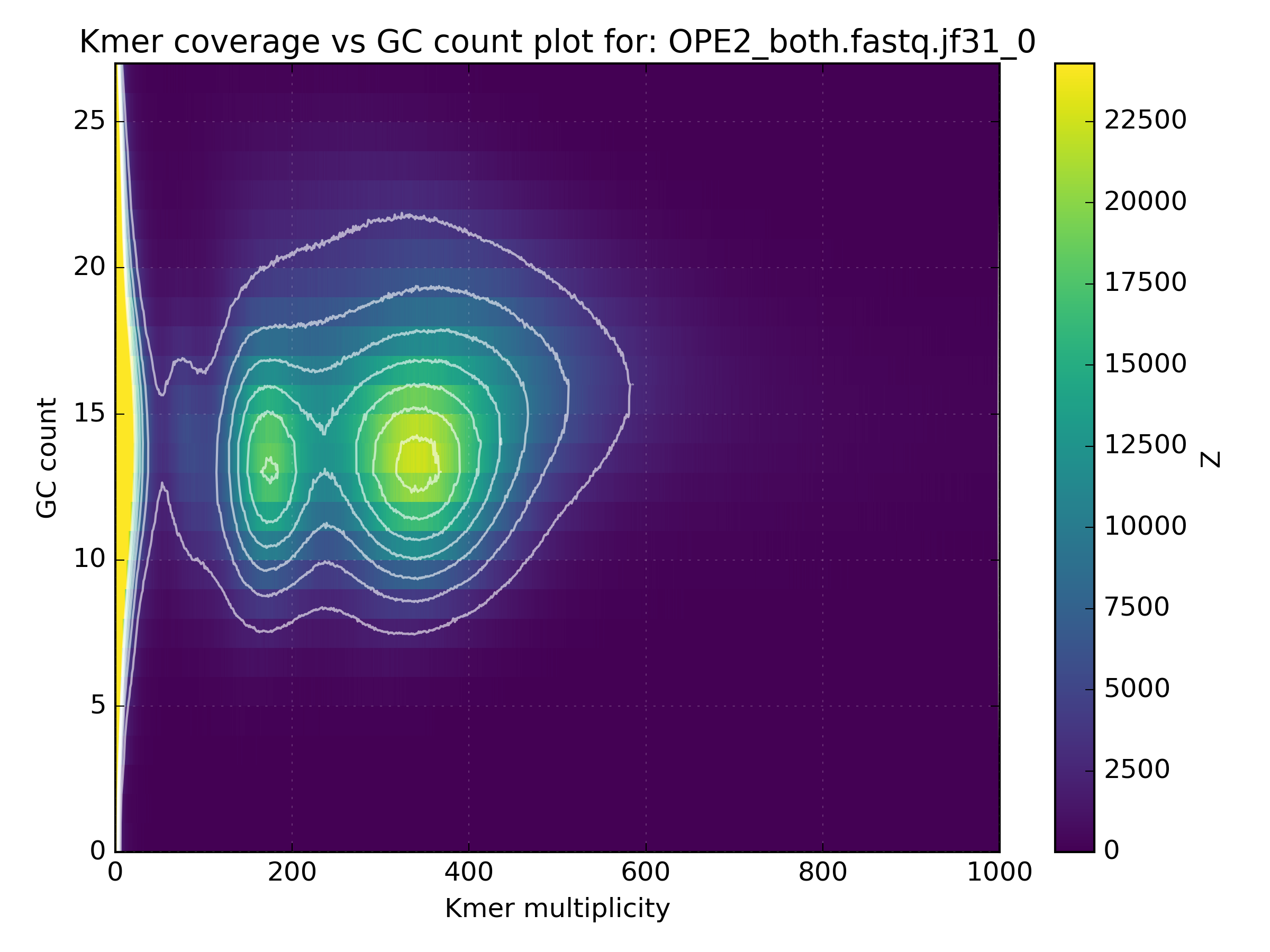
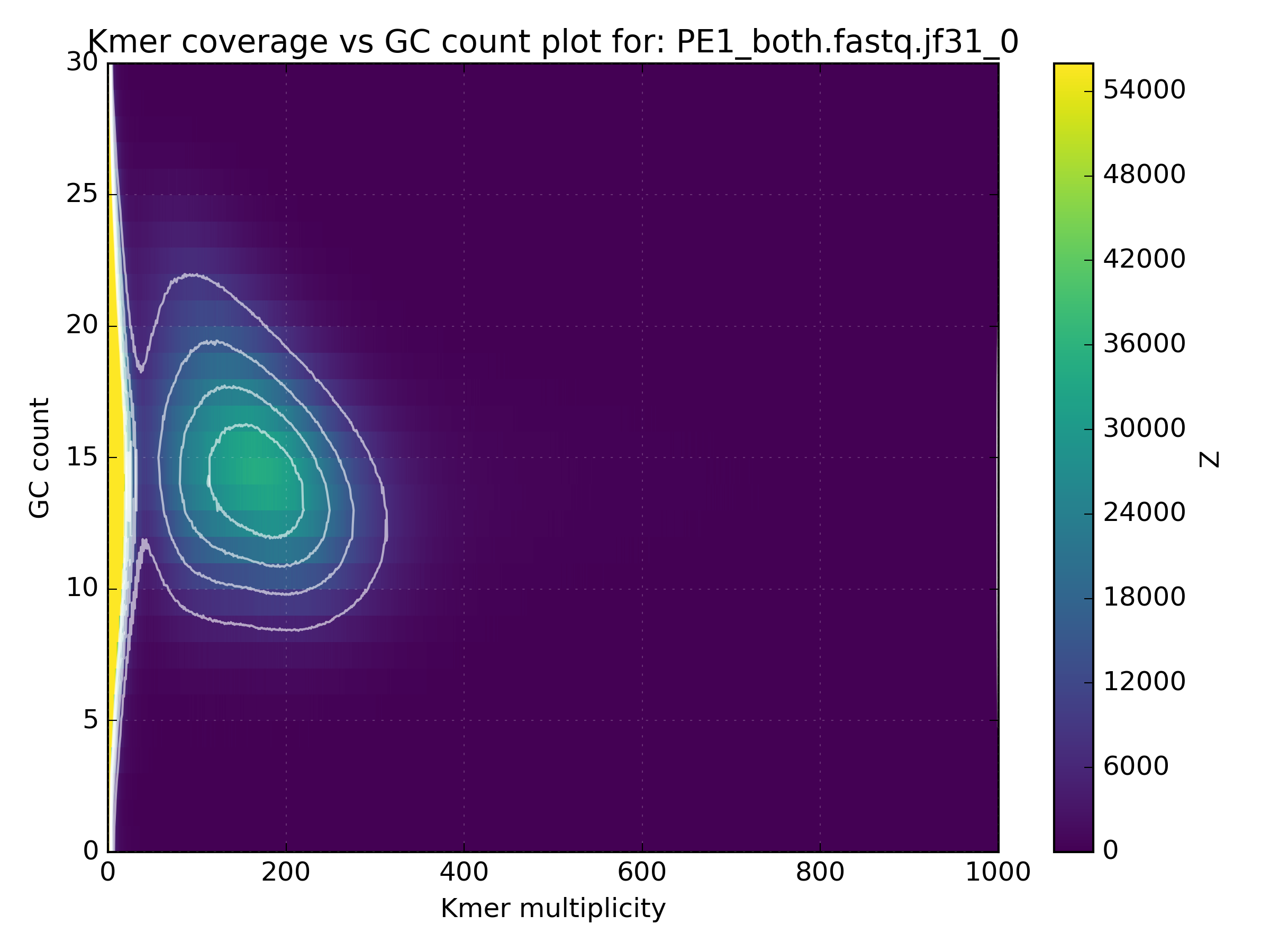
No other library contains such a hotspot at GC 15-25. After merging all libraries into one, the contaminant becomes obvious as the coverage has not altered, meaning that k-mers from this cluster were not found in the other libraries:
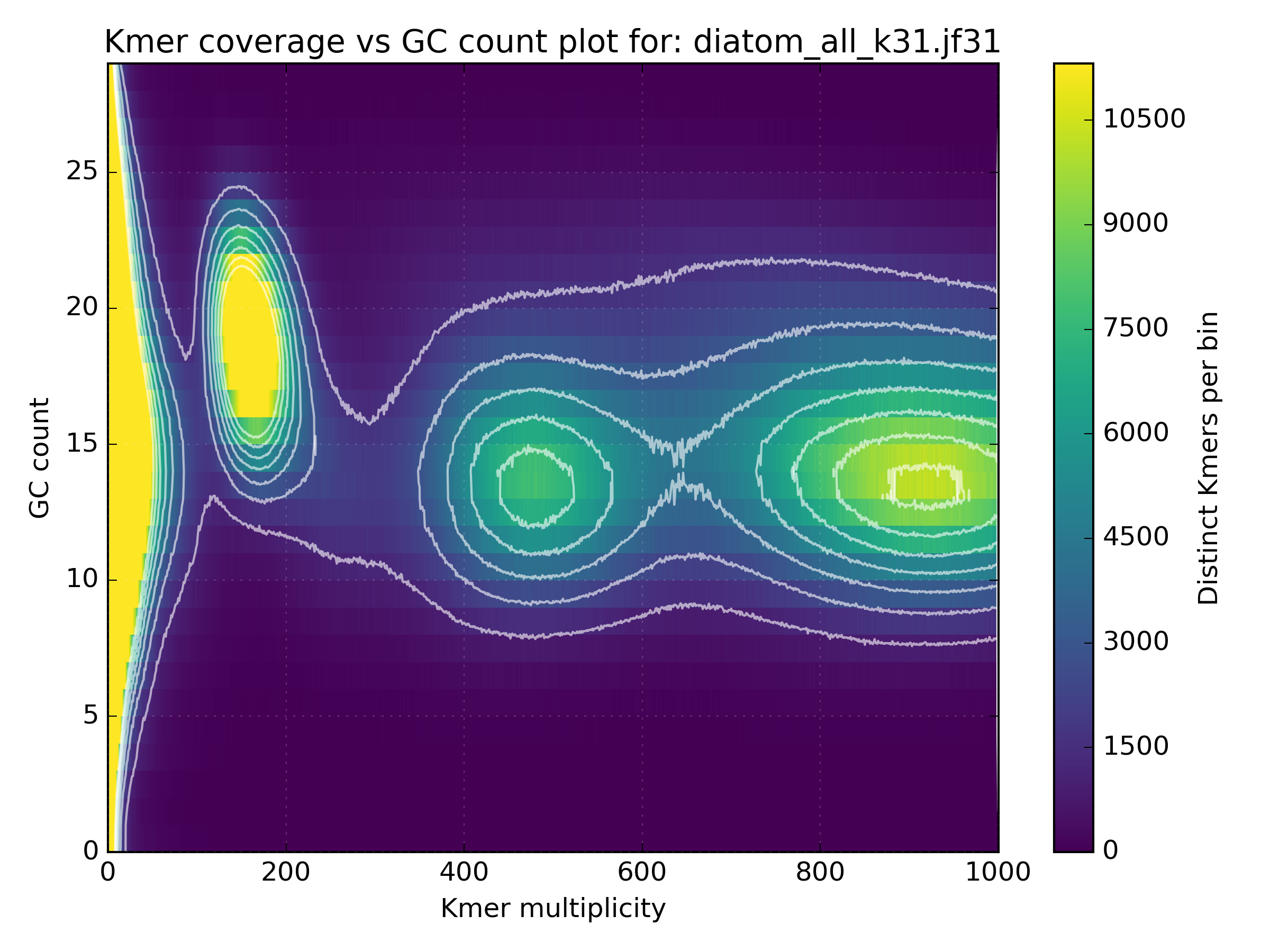
We can then use the filtering tools in KAT to extract k-mers inside, or outside defined coverage and GC limits. In this case we could take the original LMP library and run the following command:
kat filter kmer --low_count=100 --high_count=250 --low_gc=13 --high_gc=25 <path_to_MP_lib>
This produces a k-mer hash containing only those k-mer found in the defined region. We can get the reads (or assembled contigs) associated with these k-mers by running the following command:
kat filter seq --threshold=0.5 --seq=<path_to_seq_file_to_filter> --seq2=<path_to_seq_file_to_filter_2> <filtered_k-mer hash>
The example above assumes you want to filter a paired end library, although if you
want to filter single end data or and assembly you can do this by simply dropping
the --seq2 option.
BLASTing some of the sequences removed by the filtering might then identify the contaminant.
You can also use this tool for subsampling the extracted data. This can be useful
for reducing expression of highly expressed reads. To do this add the --frequency
option and set a threshold indicating how many of the reads to keep: 1.0 implies keep
all, 0.0 means discard all, 0.5 would imply to keep half of the sequences.
4.4.2. In assemblies¶
Detecting contaminants in assemblies involves a similar process to that described in the previous section. It involves marking contigs in an assembly with their average k-mer coverage and GC%.
To obtain the average coverage and GC% scores for each contig use the following command:
kat sect [options] <assembly> (<fastq>)+
By extracting the median coverage and gc% columns from the stats file it is possible to create a scatter plot which can be used in a similar way to that described in the previous section.
A second use case assumes you already know the contaminant genome and have access to the reference assembly of that contaminant. In this case you can directly inspect your assembly for signs of the contaminant using the following command:
kat sect [options] <assembly> <contaminant_genome>
This counts k-mers in the contaminant genome and applies them to the sequences in your assembly. By reverse sorting the stats file produced by the “%_non_zero_corrected” column you can identify contigs belonging to the contaminant. Normally, assuming the contaminant is the exact same species as that found in your assembly you expect to see very high percentage scores (>90%). Moderate scores (20-80%) might indicate either some shared content or chimeric sequences and should be investigated more thoroughly.
4.5. Genome assembly analysis using k-mer spectra¶
One of the most frequently used tools in KAT are the so called “assembly spectra copy number plots” or spectra-cn. We use these as a fast first analysis to check assembly coherence against the content within reads that were used to produce the assembly. Basically we represent how many elements of each frequency on the read’s spectrum ended up not included in the assembly, included once, included twice etc.
4.5.1. Homozygous genomes¶
As a simple example, we can look at how a plot for S.cerevesiae S288C would look if we are able to perfectly reconstruct the reference assembly:
kat comp -t 16 -o pe_vs_assembly 'PE.R?.fastq' assembly.fa
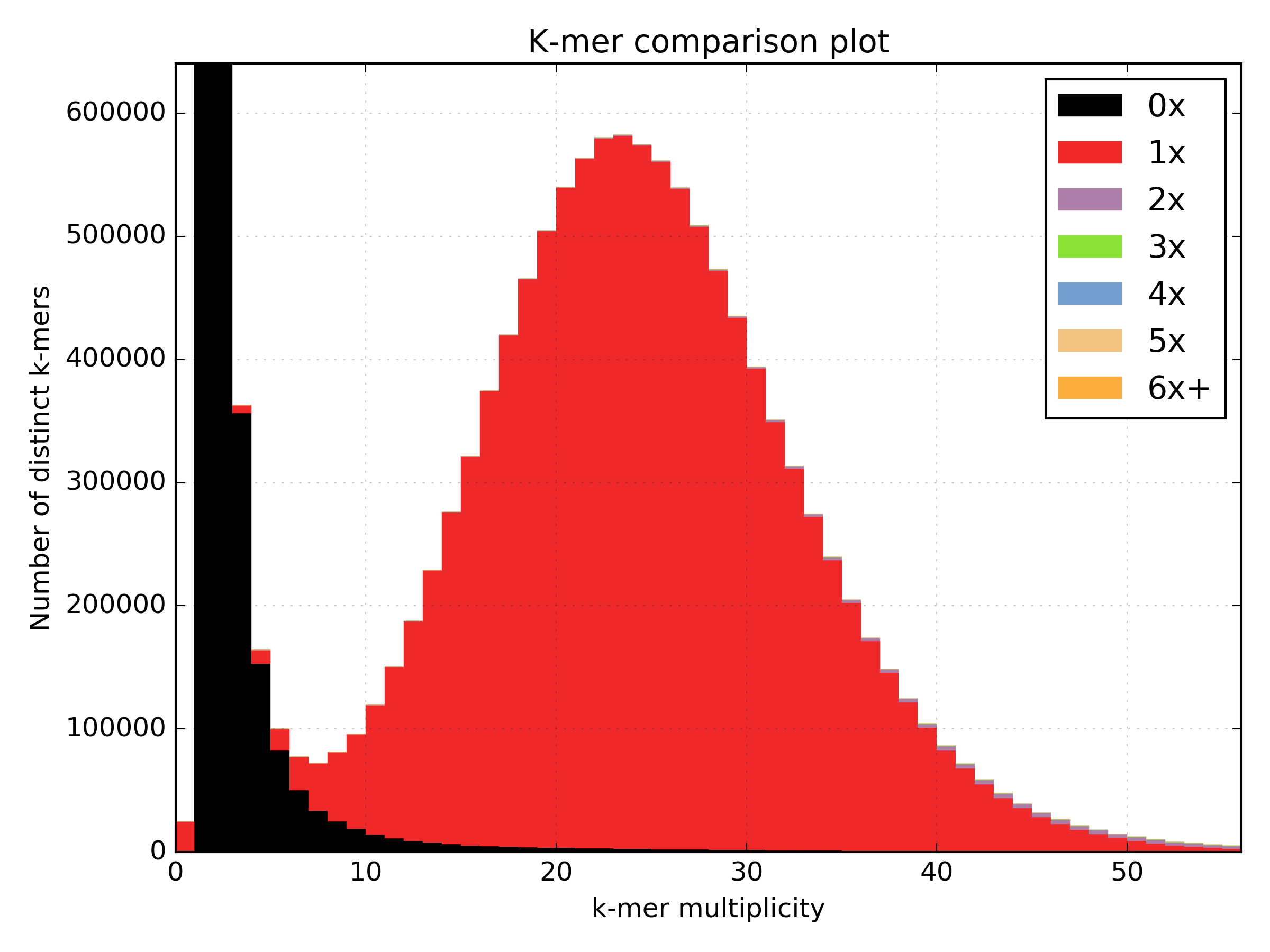
The errors are absent on the assembly, the main unique content is all there, exactly once, and all the other distributions are perfectly in place. But from the same sequencing, by choosing a wrong k-value during assembly (too small in this case), we can end up with something more interesting.
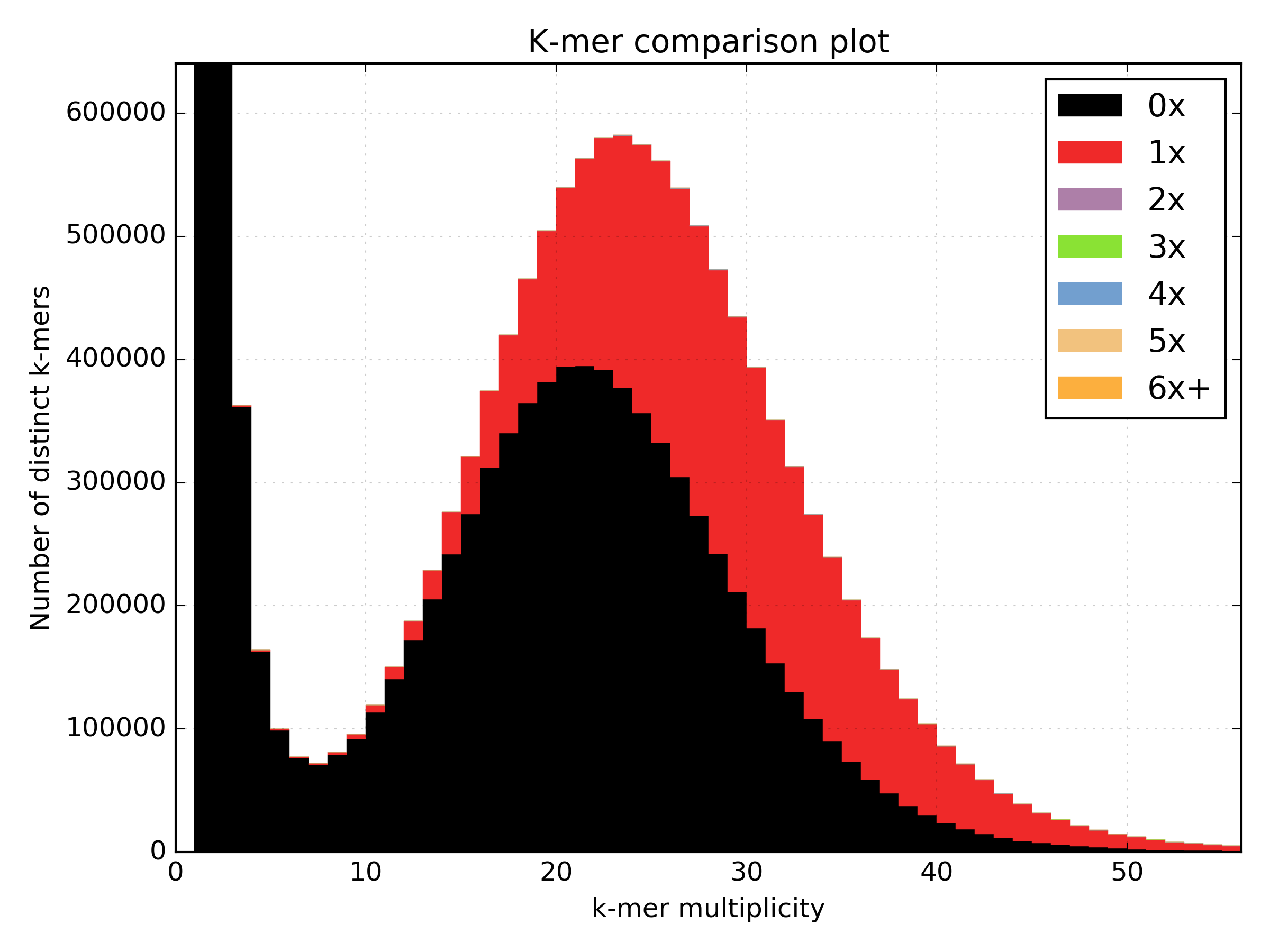
Now, in addition to the absent errors, we have a lot of missing content from the assembly.
Sometimes when we generate an assembly we want to remove short contings from the final assembly as these contigs are often not useful in downstream analysis. It is common to remove contigs shorter than 200bp, 500bp or 1Kbp but it can be a problem deciding which cutoff to use as you don’t want to remove useful content from the assembly. The spectra-cn plot is useful here as you can check assembly files (with no cutoff, 200 bp cutoff, 500bp cutoff etc.) using kat comp to quantify the content you are removing using that cutoff. Missing content is evident as a black peak below the main red peak and will increase in height as you remove more content. The choice of cutoff is a trade-off between reducing the number of contigs in the assembly and keeping as much content as possible.
4.5.2. Heterozygous genomes¶
Heterozygous genomes produce more interesting and complex plots, since the k-mer spectra clearly shows different distributions for both the heterozygous and homozygous content. The following plots show the two extremes of how a heterozygous assembly could look. The hererozygous content is represented by the first peak at x=50 and the homozygous content in the second peak at x=100. In the first plot we have a single haplotype mosaic, where the bubbles in the graph are collapsed and each heterozygous region is represented once in the assembly. This is what we typically would expect to get out of a perfect assembly. The lost content (the black peak) represents the half of the heterozygous content that is lost when bubbles are collapsed. In the second case, haplotypes are separated by duplicating all the homozygous regions and allowing us to fully capture the heterozygous content. We don’t typically, aim for the second scenario when assembling genomes.
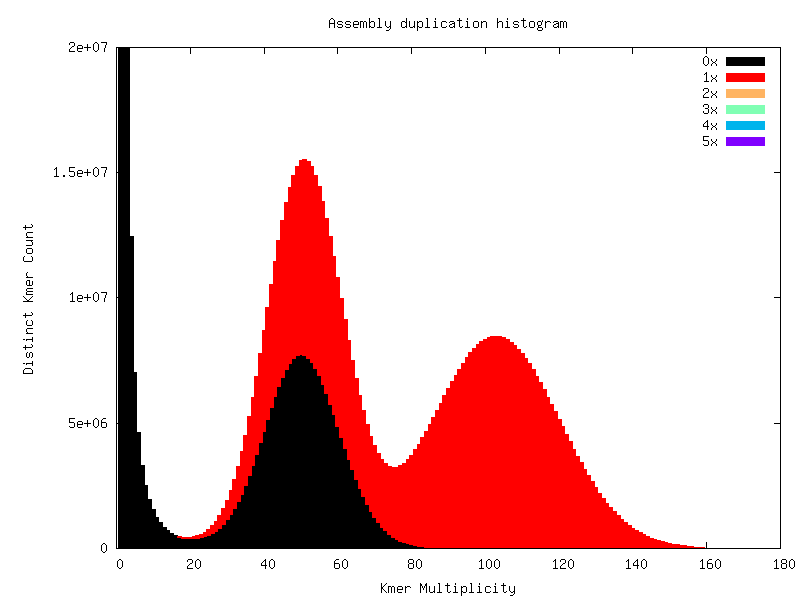
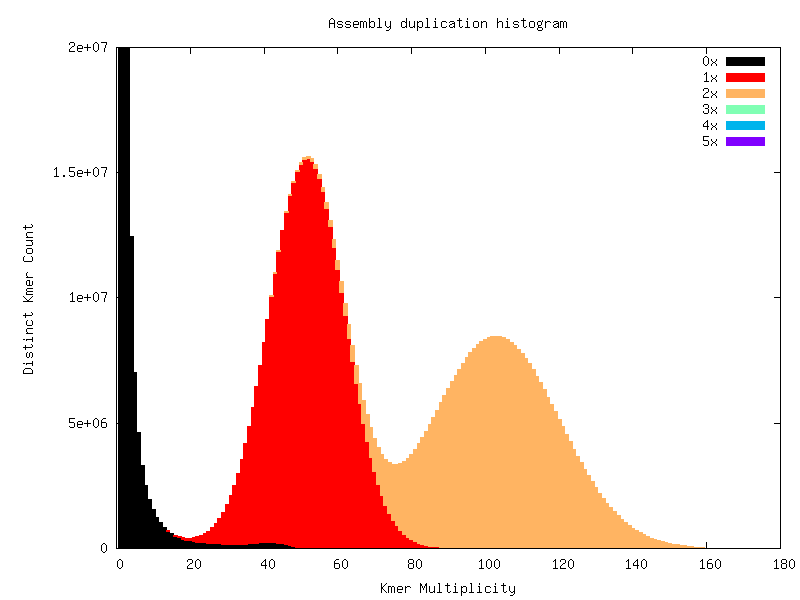
Interestingly, most assemblies don’t look like either case above but show duplications, inclusion of extra variation, etc:
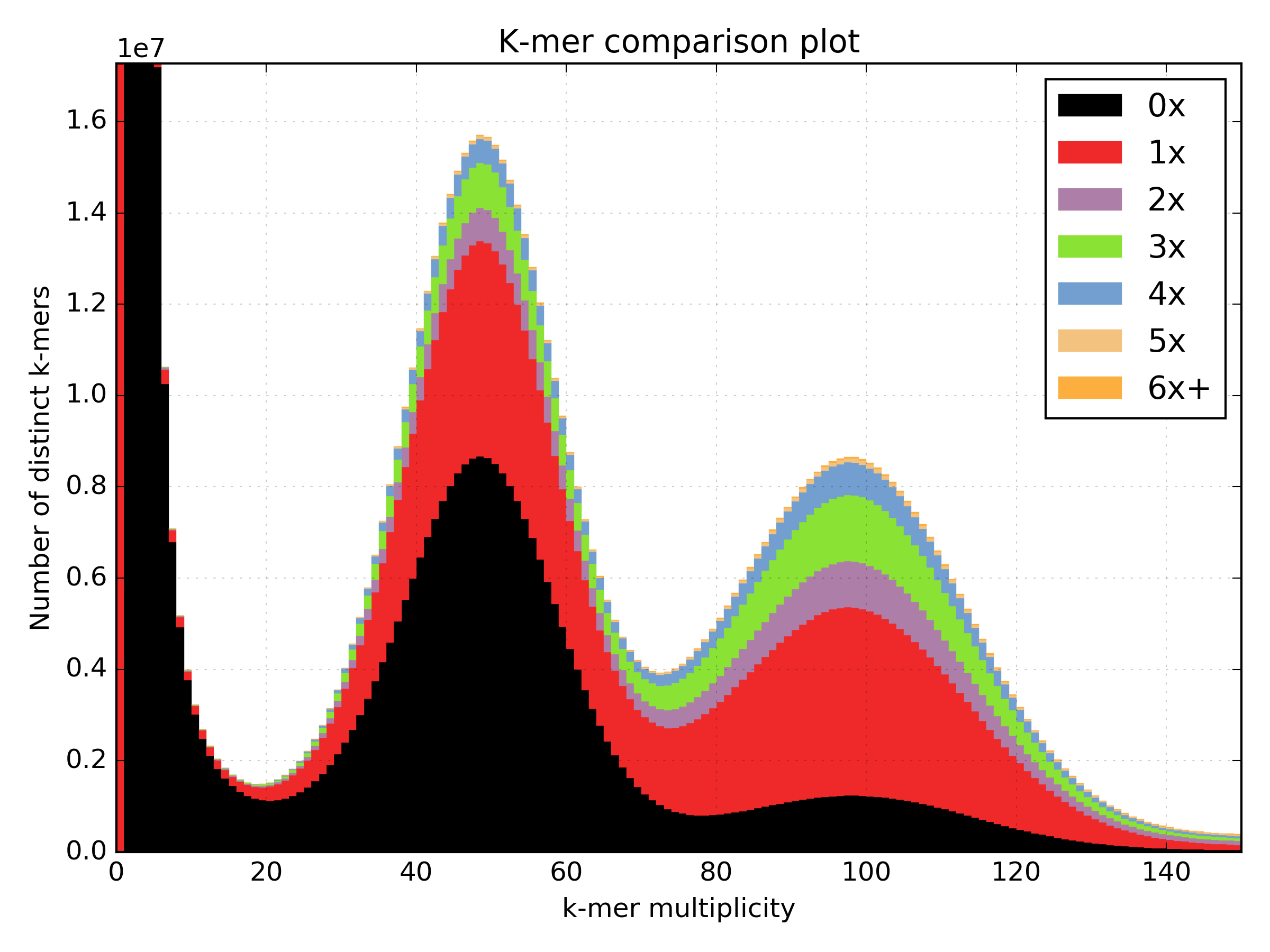
4.6. Distribution decomposition analysis¶
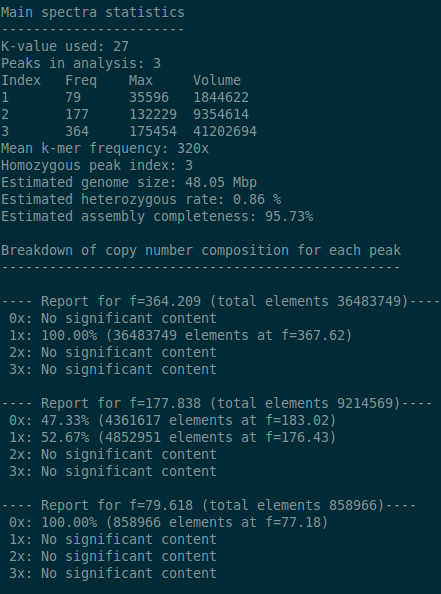
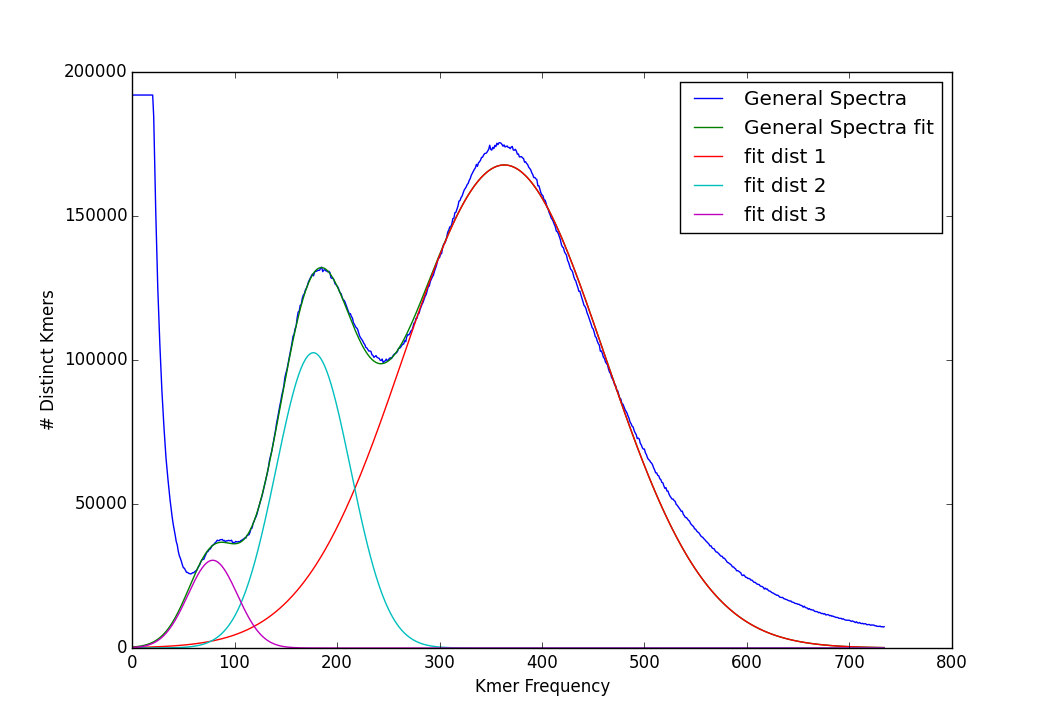
It’s useful to be able to fit distributions to each peak in a k-mer histogram, spectra-cn matrix in order to work out how many distinct k-mers can be associated with those distributions. By counting k-mers in this way we can make predictions around genome size, heterozygous rates (if diploid) and assembly completeness. To this end we bundle a script with kat called kat_distanalysis.py. It takes in either a spectra-cn matrix file, or kat histogram file as input, then proceeds to identify peaks and fit distributions to each one. In the case of spectra-cn matrix files it also identifies peaks for each copy number for an assembly. Alternatively, for matrix files generated by KAT GCP, it will also identify peaks associated with GC content. Output from this tool consists of stdout logging as well as structured JSON output. In addition, plots of the fitted distributions can be requested using the –plot option.
The user can help to get correct predictions out of the tool by providing an approximate frequency for the homozygous part of the distribution. By default, this is assumed to be the last peak. For example, this command:
kat_distanalysis.py --plot spectra-cn.mx
might produce the following output for this tetraploid genome:
As of KAT V2.4.0, this script is executed as a post-processing step to most KAT tools.
4.7. Finding repetitive regions in assemblies¶
Sometimes it’s useful to identify regions in an assembly that are repetitive. This can easily be done with the following command:
kat sect -E -F [options] <assembly_file> <assembly_file>
This counts k-mers in the assembly and then marks up the sequences in the assembly with k-mer counts at each position. Regions that have a count of 1 are extracted into a new FastA file containing non-repetitive content and regions that have a count of 2-20 (maximum threshold can be adjusted) are extracted to FastA file containing the repetitive content.